Our current and future research is focused on the development of new generation instrumentation and methodologies for biomedical and behavioral research. In particular, our group is interested in the characterization of the chemical environment at the single cell and sub-cellular level of model cell systems and tissue sections using a "molecular microscope".
There are two main challenges in the molecular characterization of native surfaces: the quantity of sample available for analysis and the dynamic mass range. The long-term goal of our work is to develop nanometer probes for mass spectrometry based imaging for the study at the single cell and sub-cellular level of "native state" biological surfaces with: i) enhanced lateral resolution, ii) enhanced emission of molecular ions and iii) fast gas-phase, post-ionization separation techniques.
This integrated approach involves the instrument development and optimization of sample-friendly conditions for the generation of the molecular ions of interest, the design and generation of MS imaging calibration procedures and standards, the incorporation of high-throughput post-ionization separation and fragmentation techniques, the analysis of the gas-phase conformational space of molecular ions, and the characterization of the chemical environment at the single cell and sub-cellular level of model biological systems.

Structural Biology Tools: HDX-TIMS-MS and molecular dynamics
Recent innovations in speed, accuracy and sensitivity have established mass spectrometry (MS) based methods as a key technology for the analysis of kinetic intermediates and folding mechanism of biological molecules and their complexes. When complemented with theoretical calculations, Ion Mobility Spectrometry – Mass Spectrometry provides a powerful tool for the identification of structural motifs. We have recently introduced Trapped Ion Mobility Spectrometry coupled to Mass Spectrometry (TIMS-MS) as a high-throughput technique for the study of solution states of biomolecules (e.g., peptides, proteins, DNA and their complexes), as well as the kinetic intermediates involved during their folding as a function of the molecular environment (e.g., pH, organic and salt content). While this description holds true for most contemporary IMS analyzers, the higher resolving power (e.g., R up to 400) and the unique ability to hold and interrogate molecular ions for kinetic studies (e.g., millisecond-second time scale) provides TIMS-MS with unique capabilities (e.g., recently combined with hydrogen-deuterium exchange, HDX-TIMS-MS and selective dopants in the TIMS cell). That is, HDX-TIMS-MS has a significant advantage in the flexibility to interrogate, at the single molecule level, the molecular interactions that define the conformational space.
Post-ionization, gas-phase separations using TIMS-FT-ICR MS/MS and TIMS-q-TOF MS/MS
My research group has special interest in the development of fast separation/identification techniques that can be easily implemented for the analysis of complex biological mixtures (e.g., gas-phase, post-ionization separation in ion mobility spectrometry and mass spectrometry). In particular, we have explored the advantages of trapped ion mobility for the analysis of complex mixtures without the need of lengthy pre-fractionation steps (e.g., GC or LC). In addition, we have developed the necessary tools for the data processing and characterization of targeted compounds using high resolution ion mobility resolution coupling to ultra-high resolution mass spectrometry.
Broader significance: The development of TIMS-FTICR MS/MS and TIMS-TOF-MS/MS has numerous applications in the characterization of complex mixtures, by reducing the sample workout, purification, preparation and preseparation steps prior MS analysis. We have shown that over a 100x fold increase in molecular identifications are possible compared to traditional separation techniques. This technology greatly reduces the cost and analysis time of complex mixtures, thus increasing the applicability of IMS-MS to biomedical, forensic and environmental problems.
Petroleomics and the analysis of fossil fuel intermediates during environmental transformation
We have recently shown that coupling fast, gas-phase post-ionization techniques (e.g., ion mobility spectrometry mass spectrometry, IMS-MS) provides an extra, orthogonal separation dimension for the analysis of fossil fuels. In particular, Ion mobility spectrometry (IMS) when combined with theoretical modeling has proven to be the most versatile technique for conformational analysis of intermediate and equilibrium structures of molecular ions by measuring the ion-neutral, collision cross-section (CCS) of molecular ions.
Broader significance: Resolving the transformation steps of fossil fuel to their molecular composition is a great challenge to analytical and environmental chemists because of the complex nature of the samples. Numerous approaches have been utilized to fully characterize fossil fuels, but no single technique is fully descriptive, and analyses by different techniques frequently generate conflicting data for the same sample. For the first time, we have been able to dissect a crude oil to their chemical composition (over 100,000 components) and to assign tridimensional structures to every component in a single analysis. The understanding of the crude oil composition and its evolution has large applications in reducing the cost of combustion processes, the treatment of the byproducts and the long term environmental effects.
Structural Biology Tools
HDX-TIMS-MS and molecular dynamics
Post-ionization, gas-phase separations using TIMS-FT-ICR MS/MS and TIMS-q-TOF MS/MS
Petroleomics and the analysis of fossil fuel intermediates during environmental transformation
Theoretical predictor for candidate structure assignment from IMS data of biomolecule-related conformational space
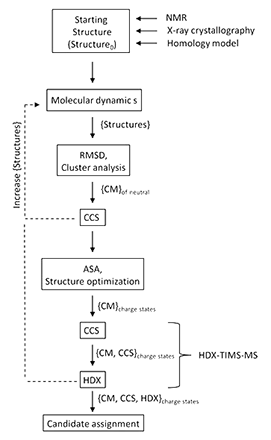
The ability to correlate experimental ion mobility data with candidate structures from theoretical modeling provides a powerful analytical and structural tool for the characterization of biomolecules. In the present paper, a theoretical workflow is described to generate and assign candidate structures for experimental trapped ion mobility and H/D exchange (HDX-TIMS-MS) data following molecular dynamics simulations and statistical filtering. The applicability of the theoretical predictor is illustrated for a peptide and protein example with multiple conformations and kinetic intermediates. The described methodology yields a low computational cost and a simple workflow by incorporating statistical filtering and molecular dynamics simulations. The workflow can be adapted to different IMS scenarios and CCS calculators for a more accurate description of the IMS experimental conditions. For the case of the HDX-TIMS-MS experiments, molecular dynamics in the “TIMS box” accounts for a better sampling of the molecular intermediates and local energy minima.
Downloads:
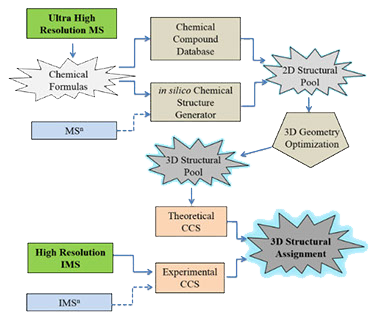
Towards unsupervised polyaromatic hydrocarbons structural assignment from
SA-TIMS–FTMS data
With the advent of high resolution ion mobility analyzers and their coupling to ultrahigh resolution mass spectrometers, there is a need to further develop a theoretical workflow capable of correlating experimental accurate mass and mobility measurements with tridimensional candidate structures. In the present work, a general workflow is de scribed for unsupervised tridimensional structural assignment based on accurate mass measurements, mobility measurements, in silico 2D-3D structure generation, and theoretical mobility calculations. In particular, the potential of this workflow will be shown for the analysis of polyaromatic hydrocarbons from Coal Tar SRM 1597a using selected accumulation - trapped ion mobility spectrometry (SA-TIMS) coupled to Fourier transform- ion cyclotron resonance mass spectrometry (FT-ICR MS). The proposed workflow can be adapted to different IMS scenarios, can utilize different collisional cross-section calculators and has the potential to include MSn and IMSn measurements for faster and more accurate tridimensional structural assignment.